Modules
flair is a wrapper script with modules for running various
processing scripts located in bin directory and should be install in your path by pip or conda.
Modules must be run in order (align, correct, collapse).
- If you want to compare multiple samples, there are two primary ways of doing this:
Combine the fastq or fasta reads of all samples and run FLAIR align, correct, and collapse on all samples together (will generate the most comprehensive transcriptome)
Run FLAIR align, correct, and collapse on each sample separately (using the –generate_map option in collapse), then combine the transcriptomes later using the collapse_bed_files script (more documentation in additional programs section)
flair align
usage: flair align -g genome.fa -r <reads.fq>|<reads.fa> [options]
This module aligns reads to the genome using minimap2, and converts the SAM output to BED12. Aligned reads in BED12 format can be visualized in IGV or the UCSC Genome browser.
Note: If you want to independently align and filter your reads and convert them to bed12, you can do so. You may want to do this if you want different alignment options - for instance, if you want to detect maximal noncanonical splice sites, you may want to align with minimap2 -un option (For this specific case to work well, you will want to run FLAIR correct with shortreads, then run collapse with –annotation_reliant and –check_splice).
Outputs
flair.aligned.bam
flair.aligned.bam.bai
flair.aligned.bed
Options
Required arguments
--reads Raw reads in fasta or fastq format. This argument accepts multiple
(comma/space separated) files.
At least one of the following arguments is required:
--genome Reference genome in fasta format. Flair will minimap index this file
unless there already is a .mmi file in the same location.
--mm_index If there already is a .mmi index for the genome it can be supplied
directly using this option.
Optional arguments
--help Show all options.
--output Name base for output files (default: flair.aligned). You can supply
an output directory (e.g. output/flair_aligned) but it has to exist;
Flair will not create it. If you run the same command twice, Flair
will overwrite the files without warning.
--threads Number of processors to use (default 4).
--junction_bed Annotated isoforms/junctions bed file for splice site-guided
minimap2 genomic alignment.
--nvrna Use native-RNA specific alignment parameters for minimap2 (-u f -k 14)
--quality Minimum MAPQ score of read alignment to the genome. The default is 1,
which is the lowest possible score.
-N Retain at most INT secondary alignments from minimap2 (default 0). Please
proceed with caution, changing this setting is only useful if you know
there are closely related homologs elsewhere in the genome. It will
likely decrease the quality of Flair's final results.
--quiet Dont print progress statements.
Notes
If you’re using human sequences, the best reference genome is GCA_000001405.15_GRCh38_no_alt_analysis_set as described in this helpful blog post by Heng Li
If your input sequences are Oxford nanopore reads, please use Pychopper before running Flair.
If your reads are already aligned, you can convert the sorted bam output to bed12 using
bam2Bed12 to supply for flair-correct. This step smoothes gaps in the alignment.
nvrna settings: See minimap2’s manual for details.
quality: More info on MAPQ scores
flair correct
usage: flair correct -q query.bed12 [-f annotation.gtf]|[-j introns.tab] -g genome.fa [options]
This module corrects misaligned splice sites using genome annotations and/or short-read splice junctions. If your genome annotation is sparse, please also use short-reads. Any reads with splice sites not near splice sites identified in orthogonal data will be thrown out.
Outputs
<args.output>_all_corrected.bedfor use in subsequent steps
<args.output>_all_inconsistent.bedrejected alignments
<args.output>_cannot_verify.bed(only if the) chromosome is not found in annotation
Options
Required arguments
--query Uncorrected bed12 file, e.g. output of flair align.
--genome Reference genome in fasta format.
At least one of the following arguments is required:
--shortread Bed format splice junctions from short-read sequencing. You can
generate these from SAM format files using the junctions_from_sam
program that comes with Flair. If you align your short reads with STAR,
you should use the SJ.out.tab file from STAR for this.
--gtf GTF annotation file.
Optional arguments
--help Show all options
--output Name base for output files (default: flair). You can supply an
output directory (e.g. output/flair) but it has to exist; Flair
will not create it. If you run the same command twice, Flair will
overwrite the files without warning.
--threads Number of processors to use (default 4).
--nvrna Specify this flag to make the strand of a read consistent with
the input annotation during correction.
--ss_window Window size for correcting splice sites (default 15).
--print_check Print err.txt with step checking.
Notes
Make sure that the genome annotation and genome sequences are compatible (if the genome sequence contains the ‘chr’ prefix, the annotations must too).
Please do use GTF instead of GFF; annotations should not split single exons into multiple entries.
flair collapse
usage: flair collapse -g genome.fa -q <query.bed> -r <reads.fq>/<reads.fa> [options]
Defines high-confidence isoforms from corrected reads. As FLAIR does not
use annotations to collapse isoforms, FLAIR will pick the name of a read
that shares the same splice junction chain as the isoform to be the
isoform name. It is recommended to still provide an annotation with
--gtf, which is used to rename FLAIR isoforms that match isoforms in
existing annotation according to the transcript_id field in the gtf.
Intermediate files generated by this step are removed by default, but
can be retained for debugging purposes by supplying the argument
--keep_intermediate and optionally supplying a directory to keep
those files with --temp_dir.
If there are multiple samples to be compared, the flair-corrected read
bed files should be concatenated prior to running
flair-collapse. In addition, all raw read fastq/fasta files should
either be specified after --reads with space/comma separators or
concatenated into a single file.
Please note: Flair collapse is not yet capable of dealing with large (>1G) input bed files. If you find that Flair needs a lot of memory you may want to follow the advice in dicussion #391 to split the bed files and reads by chromosome. We do intend to improve this.
Outputs
isoforms.bed
isoforms.gtf
isoforms.fa
If an annotation file is
provided, the isoforms ID format will contain the transcript id,
underscore, and then the gene id, so it would look like ENST*_ENSG*
if you’re working with the GENCODE human annotation.
If multiple TSSs/TESs are allowed (toggle with --max_ends or
--no_redundant), then a -1 or higher will be appended to the end
of the isoform name for the isoforms that have identical splice junction
chains and differ only by their TSS/TES.
For the gene field, the gene
that is assigned to the isoform is based on whichever annotated gene has
the greatest number of splice junctions shared with the isoform. If
there are no genes in the annotation which can be assigned to the
isoform, a genomic coordinate is used (e.g. chr*:100000).
Recommended uses
Human
The following are the recommended options to run FLAIR to increase performance on known and novel transcripts. These are the options used for submission to the Long-read RNA-Seq Genome Annotation Assessment Project systematic evaluation, which showed that FLAIR is a top-performing tool: Pardo-Palacios et al. Nature Methods 2024.
flair collapse -g genome.fa --gtf gene_annotations.gtf -q reads.flair_all_corrected.bed -r reads.fastq
--stringent --check_splice --generate_map --annotation_reliant generate
For novel isoform discovery in organisms with more unspliced transcripts and more overlapping genes, we recommend using a combination of options to capture more transcripts. For example:
Yeast
flair collapse -g genome.fa --gtf gene_annotations.gtf -q reads.flair_all_corrected.bed -r reads.fastq
--stringent --no_gtf_end_adjustment --check_splice --generate_map --trust_ends
Note that if you are doing direct-RNA, this command will likely call degradation products as isoforms. If you want to avoid this this we recommend using –annotation-reliant.
Options
Required arguments
--query Bed file of aligned/corrected reads
--genome FastA of reference genome
--reads FastA/FastQ files of raw reads, can specify multiple files
Optional arguments
--help Show all options.
--output Name base for output files (default: flair.collapse).
You can supply an output directory (e.g. output/flair_collapse)
--threads Number of processors to use (default: 4).
--gtf GTF annotation file, used for renaming FLAIR isoforms to
annotated isoforms and adjusting TSS/TESs.
--generate_map Specify this argument to generate a txt file of read-isoform
assignments (default: not specified). This file can be used to
quantify isoforms, but may produce slightly different results to
using FLAIR quantify. Also, a single read is assigned to a single isoform,
but not all reads are assigned to isoforms.
--annotation_reliant Specify transcript fasta that corresponds to transcripts
in the gtf to run annotation-reliant flair collapse; to ask flair
to make transcript sequences given the gtf and genome fa, use
--annotation_reliant generate. With this option activated, FLAIR first
aligns reads to the annotation and checks matches to annotated transcripts,
then will only identify novel transcripts from remaining reads.
Options for read support
--support Minimum number of supporting reads for an isoform; if s < 1,
it will be treated as a percentage of expression of the gene
(default: 3).
--stringent Specify if all supporting reads need to be full-length (80%
coverage and spanning 25 bp of the first and last exons).
--check_splice Enforce coverage of 4 out of 6 bp around each splice site and
no insertions greater than 3 bp at the splice site. Please note:
If you want to use --annotation_reliant as well, set it to
generate instead of providing an input transcripts fasta file,
otherwise flair may fail to match the transcript IDs.
Alternatively you can create a correctly formatted transcript
fasta file using gtf_to_bed
--trust_ends Specify if reads are generated from a long read method with
minimal fragmentation.
--quality Minimum MAPQ of read assignment to an isoform (default: 1).
Variant options
--longshot_bam BAM file from Longshot containing haplotype information for each read.
--longshot_vcf VCF file from Longshot.
For more information on the Longshot variant caller, see its github page
Transcript starts and ends
--end_window Window size for comparing transcripts starts (TSS) and ends
(TES) (default: 100).
--promoters Promoter regions bed file to identify full-length reads.
--3prime_regions TES regions bed file to identify full-length reads.
--no_redundant <none,longest,best_only> (default: none). For each unique
splice junction chain, report options include:
- none best TSSs/TESs chosen for each unique
set of splice junctions
- longest single TSS/TES chosen to maximize length
- best_only single most supported TSS/TES
--isoformtss When specified, TSS/TES for each isoform will be determined
from supporting reads for individual isoforms (default: not
specified, determined at the gene level).
--no_gtf_end_adjustment Do not use TSS/TES from the input gtf to adjust
isoform TSSs/TESs. Instead, each isoform will be determined
from supporting reads.
--max_ends Maximum number of TSS/TES picked per isoform (default: 2).
--filter Report options include:
- nosubset any isoforms that are a proper set of
another isoform are removed
- default subset isoforms are removed based on support
- comprehensive default set + all subset isoforms
- ginormous comprehensive set + single exon subset
isoforms
Other options
--temp_dir Directory for temporary files. use "./" to indicate current
directory (default: python tempfile directory).
--keep_intermediate Specify if intermediate and temporary files are to
be kept for debugging. Intermediate files include:
promoter-supported reads file, read assignments to
firstpass isoforms.
--fusion_dist Minimium distance between separate read alignments on the
same chromosome to be considered a fusion, otherwise no reads
will be assumed to be fusions.
--mm2_args Additional minimap2 arguments when aligning reads first-pass
transcripts; separate args by commas, e.g. --mm2_args=-I8g,--MD.
--quiet Suppress progress statements from being printed.
--annotated_bed BED file of annotated isoforms, required by --annotation_reliant.
If this file is not provided, flair collapse will generate the
bedfile from the gtf. Eventually this argument will be removed.
--range Interval for which to collapse isoforms, formatted
chromosome:coord1-coord2 or tab-delimited; if a range is specified,
then the --reads argument must be a BAM file and --query must be
a sorted, bgzip-ed bed file.
flair quantify
usage: flair quantify -r reads_manifest.tsv -i isoforms.fa [options]
Output
Default: only reports reads that align unambiguously to an isoform (reads that align equally to multiple isoforms are thrown out)
check_splice: adds check for read matching reference transcript at all splice sites
stringent: adds requirement for read to cover at least 25bp of the first and last exons
If you need your reads to match your isoforms well, use –check_splice and –stringent, while if you need more reads assigned to isoforms for better statistical comparison, use the default.
–quality 0 is also reccommended, as this allows slightly better recall as FLAIR can disambiguate some similar isoform alignments.
Options
Required arguments
--isoforms Fasta of Flair collapsed isoforms
--reads_manifest Tab delimited file containing sample id, condition, batch,
reads.fq, where reads.fq is the path to the sample fastq file.
Reads manifest example:
sample1 condition1 batch1 mydata/sample1.fq
sample2 condition1 batch1 mydata/sample2.fq
sample3 condition1 batch1 mydata/sample3.fq
sample4 condition2 batch1 mydata/sample4.fq
sample5 condition2 batch1 mydata/sample5.fq
sample6 condition2 batch1 mydata/sample6.fq
Note: Do not use underscores in the first three fields, see below for details.
Optional arguments
--help Show all options
--output Name base for output files (default: flair.quantify). You
can supply an output directory (e.g. output/flair_quantify).
--threads Number of processors to use (default 4).
--temp_dir Directory to put temporary files. use ./ to indicate current
directory (default: python tempfile directory).
--sample_id_only Only use sample id in output header instead of a concatenation
of id, condition, and batch.
--quality Minimum MAPQ of read assignment to an isoform (default 1).
--trust_ends Specify if reads are generated from a long read method with
minimal fragmentation.
--generate_map Create read-to-isoform assignment files for each sample.
--isoform_bed isoform .bed file, must be specified if --stringent or
--check-splice is specified.
--stringent Supporting reads must cover 80% of their isoform and extend
at least 25 nt into the first and last exons. If those exons
are themselves shorter than 25 nt, the requirement becomes
'must start within 4 nt from the start' or 'end within 4 nt
from the end'.
--check_splice Enforces coverage of 4 out of 6 bp around each splice site
and no insertions greater than 3 bp at the splice site.
--output_bam If selected, forces output of each reads file aligned to the
FLAIR transcriptome. This will be a bam with no secondary alignments
Other info
Unless --sample_id_only is specified, the output counts file concatenates id, condition and batch info for each sample. The depreciated flair_diffExp and flair_diffSplice programs expect this information.
id sample1_condition1_batch1 sample2_condition1_batch1 sample3_condition1_batch1 sample4_condition2_batch1 sample5_condition2_batch1 sample6_condition2_batch1
ENST00000225792.10_ENSG00000108654.15 21.0 12.0 10.0 10.0 14.0 13.0
ENST00000256078.9_ENSG00000133703.12 7.0 6.0 7.0 15.0 12.0 7.0
flair_diffExp
IMPORTANT NOTE: diffExp and diffSplice are not currently part of the main flair code. Instead they are supplied as separate programs named flair_diffExp and flair_diffSplice. They take the same inputs as before. These programs are deprecated and will be removed or replaced in a future release. The conda environment no long installed R and the required packages. If you find these programs useful please submit a ticket describing your needs.
usage: flair_diffExp -q counts_matrix.tsv --out_dir out_dir [options]
This module performs differential expression and differential usage analyses between exactly two conditions with 3 or more replicates. It does so by running these R packages:
If you do not have replicates you can use the diff_iso_usage standalone script.
If you have more than two sample condtions, either split your counts matrix ahead of time or run DESeq2 and DRIMSeq yourself.
Outputs
After the run, the output directory (--out_dir) contains the following, where COND1 and COND2 are the names of the sample groups.
genes_deseq2_MCF7_v_A549.tsvFiltered differential gene expression table.
genes_deseq2_QCplots_MCF7_v_A549.pdfQC plots, see the DESeq2 manual for details.
isoforms_deseq2_MCF7_v_A549.tsvFiltered differential isoform expression table.
isoforms_deseq2_QCplots_MCF7_v_A549.pdfQC plots
isoforms_drimseq_MCF7_v_A549.tsvFiltered differential isoform usage table
workdirTemporary files including unfiltered output files.
Options
Required arguments
--counts_matrix Tab-delimited isoform count matrix from flair quantify
--out_dir Output directory for tables and plots.
Optional arguments
--help Show this help message and exit
--threads Number of threads for parallel DRIMSeq.
--exp_thresh Read count expression threshold. Isoforms in which both
conditions contain fewer than E reads are filtered out (Default E=10)
--out_dir_force Specify this argument to force overwriting of files in
an existing output directory
Notes
DESeq2 and DRIMSeq are optimized for short read experiments and expect many reads for each expressed gene. Lower coverage (as expected when using long reads) will tend to result in false positives.
For instance, look at this counts table with two groups (s and v) of three samples each:
gene s1 s2 s3 v1 v2 v3
A 1 0 2 0 4 2
B 100 99 101 100 104 102
Gene A has an average expression of 1 in group s, and 2 in group v but the total variation in read count is 0-4. The same variation is true for gene B, but it will not be considered differentially expressed.
Flair does not remove low count genes as long as they are expressed in all samples of at least one group so please be careful when interpreting results.
Results tables are filtered and reordered by p-value so that only p<0.05 differential genes/isoforms remain. Unfiltered tables can be found in workdir
Code requirements
This module requires python modules and R packages that are not necessary for other Flair modules (except diffSplice). You must install R” and these packages to use `diffExp and diffSplice:
flair diffSplice
IMPORTANT NOTE: diffExp and diffSplice are not currently part of the main flair code. Instead they are supplied as separate programs named flair_diffExp and flair_diffSplice. They take the same inputs as before. These programs are deprecated and will be removed or replaced in a future release. The conda environment no long installed R and the required packages. If you find these programs useful please submit a ticket describing your needs.
usage: flair_diffSplice -i isoforms.bed -q counts_matrix.tsv [options]
This module calls alternative splicing (AS) events from isoforms. Currently supports the following AS events:
intron retention (ir)
alternative 3’ splicing (alt3)
alternative 5’ splicing (alt5)
cassette exons (es)
If there are 3 or more samples per condition, then you can run with
--test and DRIMSeq will be used to calculate differential usage of
the alternative splicing events between two conditions. See below for
more DRIMSeq-specific arguments.
If conditions were sequenced without replicates, then the diffSplice output files can be input to the diffsplice_fishers_exact script for statistical testing instead.
Outputs
After the run, the output directory (--out_dir) contains the following tab separated files:
diffsplice.alt3.events.quant.tsv
diffsplice.alt5.events.quant.tsv
diffsplice.es.events.quant.tsv
diffsplice.ir.events.quant.tsv
If DRIMSeq was run (where A and B are conditionA and conditionB, see below):
drimseq_alt3_A_v_B.tsv
drimseq_alt5_A_v_B.tsv
drimseq_es_A_v_B.tsv
drimseq_ir_A_v_B.tsv
workdirTemporary files including unfiltered output files.
Options
Required arguments
--isoforms Isoforms in bed format from Flair collapse.
--counts_matrix Tab-delimited isoform count matrix from Flair quantify.
--out_dir Output directory for tables and plots.
Optional arguments
--help Show all options.
--threads Number of processors to use (default 4).
--test Run DRIMSeq statistical testing.
--drim1 The minimum number of samples that have coverage over an
AS event inclusion/exclusion for DRIMSeq testing; events
with too few samples are filtered out and not tested (6).
--drim2 The minimum number of samples expressing the inclusion of
an AS event; events with too few samples are filtered out
and not tested (3).
--drim3 The minimum number of reads covering an AS event
inclusion/exclusion for DRIMSeq testing, events with too
few samples are filtered out and not tested (15).
--drim4 The minimum number of reads covering an AS event inclusion
for DRIMSeq testing, events with too few samples are
filtered out and not tested (5).
--batch If specified with --test, DRIMSeq will perform batch correction.
--conditionA Specify one condition corresponding to samples in the
counts_matrix to be compared against condition2; by default,
the first two unique conditions are used. This implies --test.
--conditionB Specify another condition corresponding to samples in the
counts_matrix to be compared against conditionA.
--out_dir_force Specify this argument to force overwriting of files in an
existing output directory
Notes
Results tables are filtered and reordered by p-value so that only p<0.05 differential genes/isoforms remain. Unfiltered tables can be found in workdir
For a complex splicing example, please note the 2 alternative 3’ SS, 3
intron retention, and 4 exon skipping events in the following set of
isoforms that flair diffSplice would call and the isoforms that are
considered to include or exclude the each event:
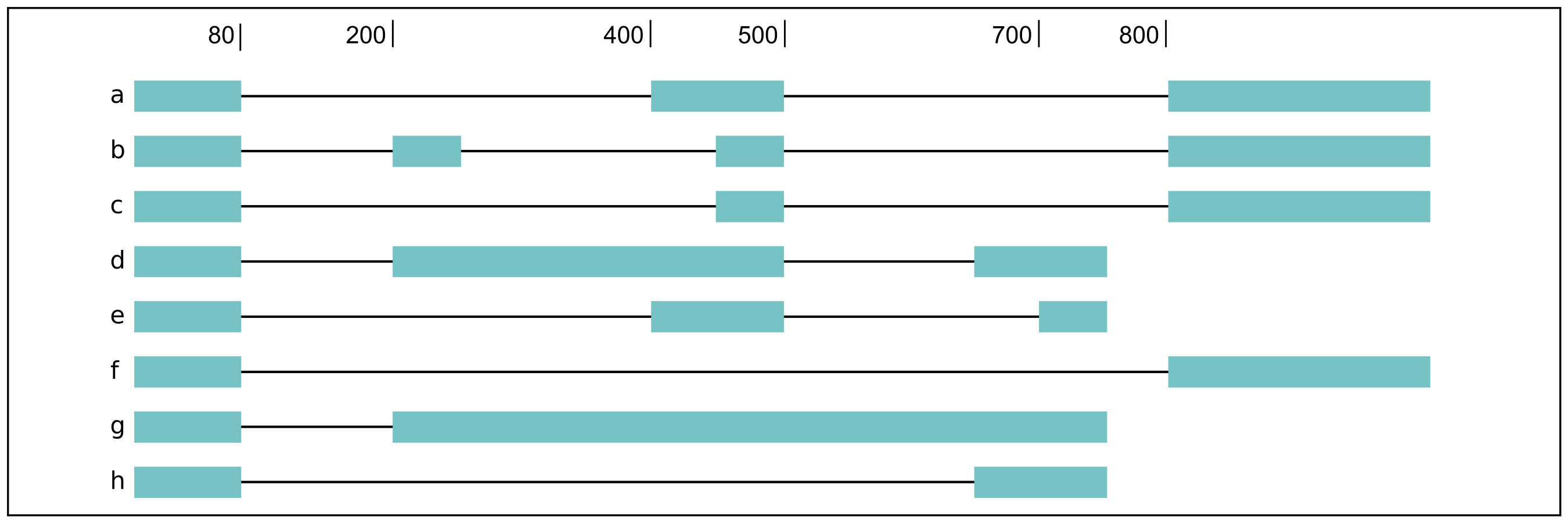
a3ss_feature_id coordinate sample1 sample2 ... isoform_ids
inclusion_chr1:80 chr1:80-400_chr1:80-450 75.0 35.0 ... a,e
exclusion_chr1:80 chr1:80-400_chr1:80-450 3.0 13.0 ... c
inclusion_chr1:500 chr1:500-650_chr1:500-700 4.0 18.0 ... d
exclusion_chr1:500 chr1:500-650_chr1:500-700 70.0 17.0 ... e
ir_feature_id coordinate sample1 sample2 ... isoform_ids
inclusion_chr1:500-650 chr1:500-650 46.0 13.0 ... g
exclusion_chr1:500-650 chr1:500-650 4.0 18.0 ... d
inclusion_chr1:500-700 chr1:500-700 46.0 13.0 ... g
exclusion_chr1:500-700 chr1:500-700 70.0 17.0 ... e
inclusion_chr1:250-450 chr1:250-450 50.0 31.0 ... d,g
exclusion_chr1:250-450 chr1:250-450 80.0 17.0 ... b
es_feature_id coordinate sample1 sample2 ... isoform_ids
inclusion_chr1:450-500 chr1:450-500 83.0 30.0 ... b,c
exclusion_chr1:450-500 chr1:450-500 56.0 15.0 ... f
inclusion_chr1:200-250 chr1:200-250 80.0 17.0 ... b
exclusion_chr1:200-250 chr1:200-250 3.0 13.0 ... c
inclusion_chr1:200-500 chr1:200-500 4.0 18.0 ... d
exclusion_chr1:200-500 chr1:200-500 22.0 15.0 ... h
inclusion_chr1:400-500 chr1:400-500 75.0 35.0 ... e,a
exclusion_chr1:400-500 chr1:400-500 56.0 15.0 ... f