Additional programs
When you conda install flair, the following helper programs will be in your $PATH:
collapse_bed_files
- usage: flair_combine [-h] -m MANIFEST [-o OUTPUT_PREFIX] [-w ENDWINDOW]
[-p MINPERCENTUSAGE] [-c] [-s] [-f FILTER]
- options:
- -h, --help
show this help message and exit
- -m MANIFEST, --manifest MANIFEST
path to manifest files that points to transcriptomes to combine. Each line of file should be tab separated with sample name, sample type (isoform or fusionisoform), path/to/isoforms.bed, path/to/isoforms.fa, path/to/combined.isoform.read.map.txt. fa and read.map.txt files are not required, although if .fa files are not provided for each sample a .fa output will not be generated
- -o OUTPUT_PREFIX, --output_prefix OUTPUT_PREFIX
path to collapsed_output.bed file. default: ‘collapsed_flairomes’
- -w ENDWINDOW, --endwindow ENDWINDOW
window for comparing ends of isoforms with the same intron chain. Default:200bp
- -p MINPERCENTUSAGE, --minpercentusage MINPERCENTUSAGE
minimum percent usage required in one sample to keep isoform in combined transcriptome. Default:10
- -c, --convert_gtf
[optional] whether to convert the combined transcriptome bed file to gtf
- -s, --include_se
whether to include single exon isoforms. Default: dont include
- -f FILTER, --filter FILTER
type of filtering. Options: usageandlongest(default), usageonly, none, or a number for the total count of reads required to call an isoform
Combines FLAIR transcriptomes or with other FLAIR transcriptomes or annotation transcriptomes to generate accurate combined transcriptome. Only the manifest file is required. Manifest file is in the following format:
sample1 isoform sample1.FLAIR.isoforms.bed sample1.FLAIR.isoforms.fa sample1.FLAIR.isoforms.fa sample1.read.map.txt
sample2 isoform sample2.FLAIR.isoforms.bed sample2.FLAIR.isoforms.fa sample2.FLAIR.isoforms.fa sample2.read.map.txt
For each line, the sample name and bed path is required. The fasta and read.map.txt file is optional. Without these files there is less ability to filter and more isoforms will be included. If a sample is a FLAIR run, we highly recommend including the read.map.txt file. If you want to combine FLAIR transcriptomes with annotated transcripts, you can convert an annotation gtf file to a bed file using
diff_iso_usage
usage: diff_iso_usage counts_matrix colname1 colname2 diff_isos.txt
Calculates the usage of each isoform as a fraction of the total expression of the gene and compares this between samples.
Requires four positional arguments to identify and calculate
significance of alternative isoform usage between two samples using
Fisher’s exact tests: (1) counts_matrix.tsv from flair-quantify, (2) the
name of the column of the first sample, (3) the name of the column of
the second sample, (4) txt output filename containing the p-value
associated with differential isoform usage for each isoform. The more
differentially used the isoforms are between the first and second
condition, the lower the p-value.
Output file format columns are as follows:
gene name
isoform name
p-value
sample1 isoform count
sample2 isoform count
sample1 alternative isoforms for gene count
sample2 alternative isoforms for gene count
diffsplice_fishers_exact
usage: diffsplice_fishers_exact events.quant.tsv colname1 colname2 out.fishers.tsv
Identifies and calculates the significance of alternative splicing
events between two samples without replicates using Fisher’s exact
tests. Requires four positional arguments: (1) flair-diffSplice tsv
of alternative splicing calls for a splicing event type, (2) the name of
the column of the first sample, (3) the name of the column of the second
sample, and (4) tsv output filename containing the p-values from
Fisher’s exact tests of each event.
Output
The output file contains the original columns with an additional column containing the p-values appended.
fasta_seq_lengths
usage: fasta_seq_lengths fasta outfilename [outfilename2]
junctions_from_sam
Usage: junctions_from_sam [options]
Options:
-h, --help show this help message and exit
-s SAM_FILE SAM/BAM file of read alignments to junctions and
the genome. More than one file can be listed,
but comma-delimited, e.g file_1.bam,file_2.bam
--unique Only keeps uniquely aligned reads. Looks at NH
tag to be 1 for this information.
-n NAME Name prefixed used for output BED file.
Default=junctions_from_sam
-l READ_LENGTH Expected read length if all reads should be of
the same length
-c CONFIDENCE_SCORE The mininmum entropy score a junction
has to have in order to be considered
confident. The entropy score =
-Shannon Entropy. Default=1.0
-j FORCED_JUNCTIONS File containing intron coordinates
that correspond to junctions that will be
kept regardless of the confidence score.
-v Will run the program with junction strand ambiguity
messages
mark_intron_retention
usage: mark_intron_retention in.bed out_isoforms.bed out_introns.txt
Assumes the bed has the correct strand information
Requires three positional arguments to identify intron retentions in isoforms:
in.bedBED of isoforms
out_isoforms.bedoutput filename
out_introns.txtoutput filename for coordinates of introns found.
Outputs
an extended
BEDwith an additional column containing either values 0 or 1 classifying the isoform as either spliced or intron-retaining, respectively
txtfile of intron retentions with formatisoform namechromosomeintron 5' coordinateintron 3' coordinate.
Note: A bed file with more additional columns will not be displayed in the UCSC genome browser, but can be displayed in IGV.
mark_productivity
usage: mark_productivity reads.psl annotation.gtf genome.fa > reads.productivity.psl
normalize_counts_matrix
usage: normalize_counts_matrix matrix outmatrix [cpm/uq/median] [gtf]
Gtf if normalization by protein coding gene counts only
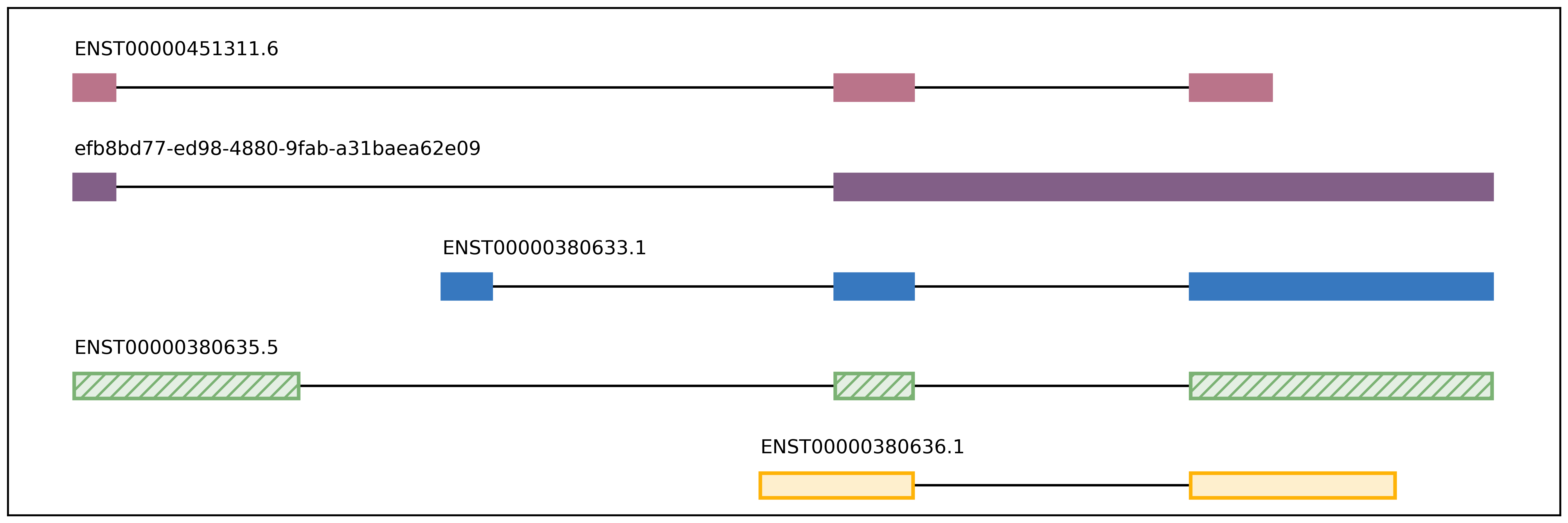
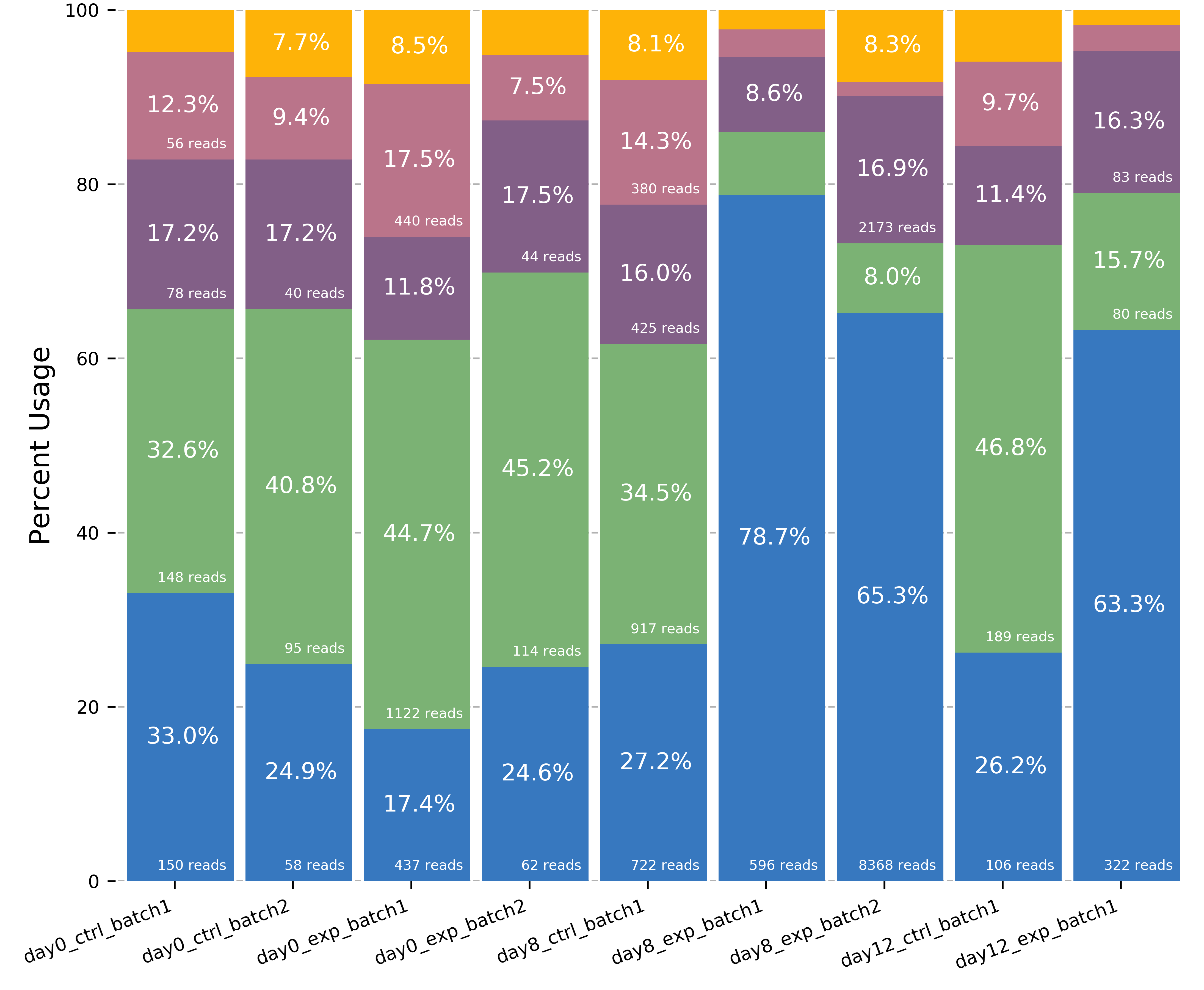
plot_isoform_usage
plot_isoform_usage <isoforms.bed> counts_matrix.tsv gene_name
Visualization script for FLAIR isoform structures and the percent usage
of each isoform in each sample for a given gene. If you supply the
isoforms.bed file from running predictProductivity, then isoforms
will be filled according to the predicted productivity (solid for
PRO, hatched for PTC, faded for NGO or NST). The gene
name supplied should correspond to a gene name in your isoform file and
counts file.
The script will produce two images, one of the isoform models and another of the usage proportions.
The most highly expressed isoforms across all the samples will be plotted.
The minor isoforms are aggregated into a gray bar. You can toggle min_reads or color_palette to plot more isoforms. Run with –help for options
Outputs
gene_name_isoforms.png of isoform structures
gene_name_usage.png of isoform usage by sample
For example:
positional arguments:
isoforms isoforms in bed format
counts_matrix genomic sequence
gene_name Name of gene, must correspond with the gene names in
the isoform and counts matrix files
options:
-h, --help show this help message and exit
-o O prefix used for output files (default=gene_name)
--min_reads MIN_READS
minimum number of total supporting reads for an
isoform to be visualized (default=6)
-v VCF, --vcf VCF VCF containing the isoform names that include each
variant in the last sample column
--palette PALETTE provide a palette file if you would like to visualize
more than 7 isoforms at once or change the palette
used. each line contains a hex color for each isoform
predictProductivity
usage: predictProductivity -i isoforms.bed -f genome.fa -g annotations.gtf
Annotated start codons from the annotation are used to identify the
longest ORF for each isoform for predicting isoform productivity.
Requires three arguments to classify isoforms according to productivity:
(1) isoforms in bed format, (2) gtf genome
annotation, (3) fasta genome sequences. Bedtools must be in your
$PATH for predictProductivity to run properly.
Output
Outputs a bed file with either the values PRO (productive), PTC
(premature termination codon, i.e. unproductive), NGO (no start
codon), or NST (has start codon but no stop codon) appended to the
end of the isoform name. When isoforms are visualized in the UCSC genome
browser or IGV, the isoforms will be colored accordingly and have
thicker exons to denote the coding region.
options:
-h, --help show this help message and exit
-i INPUT_ISOFORMS, --input_isoforms INPUT_ISOFORMS
Input collapsed isoforms in bed12 format.
-g GTF, --gtf GTF Gencode annotation file.
-f GENOME_FASTA, --genome_fasta GENOME_FASTA
Fasta file containing transcript sequences.
--quiet Do not display progress
--append_column Append prediction as an additional column in file
--firstTIS Defined ORFs by the first annotated TIS.
--longestORF Defined ORFs by the longest open reading frame.
File conversion scripts
bam2Bed12
usage: bam2Bed12 -i sorted.aligned.bam
options:
-h, --help show this help message and exit
-i INPUT_BAM, --input_bam Input bam file.
--keep_supplementary Keep supplementary alignments
A tool to convert minimap2 BAM to Bed12.
sam_to_map
usage: sam_to_map sam outfile