Welcome to FLAIR’s documentation!
Staying in touch and getting help
Please subscribe to the FLAIR announce mailing list at:
FLAIR Announce Group
This is a read-only, low volume list that will only have announcements of new FLAIR releases, publications, and other FLAIR-related user information.
If you have questions, please ask in FLAIR GitHub Discussions:
Report bugs to FLAIR GitHub Issues:
Installing FLAIR
FLAIR can be conda installed using
conda create -n flair -c conda-forge -c bioconda flair
conda activate flair
On Apple Silicon Mac systems (ARM64 CPUs: M1, M2, …) you must use
CONDA_SUBDIR=osx-64 conda create -n flair
conda activate flair
conda config --env --set subdir osx-64
conda config --add channels bioconda
conda config --add channels conda-forge
conda install flair
Note that mamba currently fails to install FLAIR on Mac ARM64.
FLAIR can be run optionally with short-read data to help increase splice site accuracy of the long read splice junctions. FLAIR uses multiple alignment steps and splice site filters to increase confidence in the set of isoforms defined from noisy data. FLAIR was designed to be able to sense subtle splicing changes in nanopore data from Tang et al. (2020). Please read for more description of the methods.
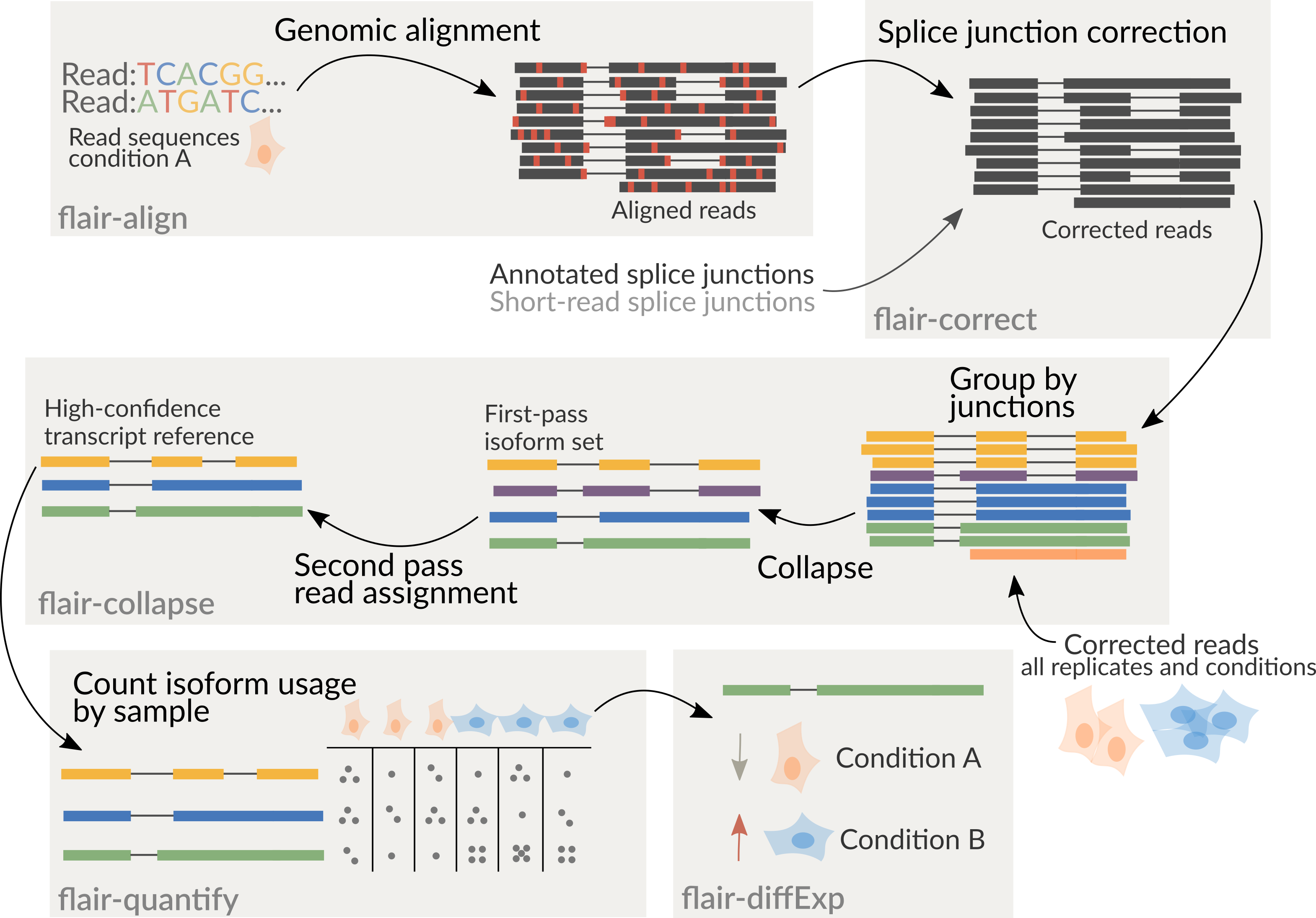
If you have multiple samples and want to compare them on a single transcriptome, you have two options:
Run flair correct and collapse individually on each sample, then combine your
transcriptomes using collapse_bed_files (see Additional programs). This method
will be faster and easier, but you may miss some low-expression transcripts.
Your other option is to merge your samples before running FLAIR. If using PacBio reads, be careful doing this, as the reads may not have unique IDs. You may need to label each read with its sample ID to keep the read IDs unique. You can either merge the FASTA/FASTQ files before running FLAIR (simplest, recommended), or merge the bed files after running FLAIR correct, making sure to run FLAIR collapse with a list of all of your read files.
It is recommended to combine all samples together prior to running
flair-collapse for isoform assembly by concatenating corrected read bed
files together. Following the creation of an isoform reference from
flair-collapse, consequent steps will assign reads from each sample
individually to isoforms of the combined assembly for downstream analyses.